

IP 501 Determination of Aluminium, Silicon, Vanadium, Nickel, Iron, Sodium, Calcium, Zinc and Phosphorus in Residual Fuel Oil by Ashing, Fusion and Inductively Coupled Plasma Emission Spectrometry
7 Procedure
7.1 Test portion mass
Select a mass of test portion, between 20 g and 50 g, to yield approximately 5 - 50 mg of ash.
NOTE 3 - The mass of test portion is selected based on the ash content. If the levels of any elements is outside the validated calibration range, dilution of the test solutions are required.
7.2 Test solution preparation
7.2.1 Immediately after homogenization, transfer the test portion from the container to the weighed platinum basin (5.3). Re-weigh the basin and contents to the nearest 0,1 g to obtain the test portion mass.
7.2.2 If required, add 0,3 g of ashing agent (4.3) to the basin, distributing it as evenly as possible over the surface of the basin contents.
If only aluminium, silicon and iron are being determined, or if it is known that the ashing agent being used does not contribute significantly to the background blank, continue directly with the procedure described in 7.2.3 onwards. If however, the ashing agent has an unknown background effect, or if vanadium, nickel, sodium, calcium, zinc and/or phosphorus is to be included in the determination, prepare a blank test portion containing only the ashing agent, and use it for the complete procedure in this clause.
7.2.3 Warm the basin and contents gently with a Bunsen flame until the test portion ignites. Maintain the contents of the basin at a temperature such that most of the combustible material is removed and only carbon and ash remain.
NOTE 4 - If the sample contains considerable amounts of moisture, foaming and frothing may cause loss of test portion.
If foaming and frothing occur, discard the test portion and add 1 ml to 2 ml of propan-2-ol (4.4) before heating. If foaming and frothing are not sufficiently reduced, add 10 ml of toluene/propan-2-ol mixture (4.6) to a further test portion and mix thoroughly. Place several strips of filter paper (5.14) in the mixture before warming gently.
NOTE 5 - When the paper begins to burn, the greater part of the water will have been removed.
7.2.4 Place the basin and its contents in the muffle furnace (5.7), preheated to a temperature of 525 °C±25 °C. Ensure that the basin contents are not contaminated with refractory from the furnace walls, as this will contribute to erroneous silicon results. Maintain the muffle furnace at this temperature until all the carbon has been removed and only ash remains.
NOTE 6 - This may take overnight.
7.2.5 Cool the basin to room temperature, add 0,4 g of flux (4.8), and mix it with the ash. Place the basin and contents in the muffle furnace preheated to a temperature of 925 °C±25 °C for 5 min. Remove the basin and ensure contact of the flux with the ash. Replace the basin in the muffle furnace and maintain the temperature at 925 °C±25 °C for a further 10 min.
7.2.6 Remove the basin and cool the fusion melt to room temperature. Add 50 ml tartaric acid/hydrochloric acid solution (4.9.1) to the basin and place on the hot plate (5.8). Maintain at a moderate temperature without boiling.
NOTE 7 - Excessive evaporation of the solution could lead to precipitation of an insoluble form of silica.
NOTE 8 - Prolonged heating may be required to dissolve the solidified melt completely and obtain a solution. Agitation, or the use of magnetic stirring may be employed to speed dissolution of the solidified melt.
7.2.7 Allow the solution to cool and then transfer it to a 100 ml volumetric flask (5.9) with water, washing the basin several times to ensure that the transfer is quantitative. Make up to the mark with water, and transfer immediately to a 100 ml plastic bottle (5.15).
NOTE 9 - Transfer to a plastic bottle is recommended as the dilute acid solution will contain tetrafluoroboric acid (HBF4) from solution of flux. However, storage tests have shown that there is no attack of glassware in the short term (up to one week), and that the solution does not contain free fluoride 'ion' above the 5 mg/l level.
7.3 Preparation of blank solution
Prepare a blank solution containing only 0,4 g flux (4.8) and 50 ml of the tartaric acid/hydrochloric acid solution (4.9.1), diluted to 100 ml, and transfer immediately to a 100 ml plastic bottle (5.15).
7.4 Preparation of calibration solutions
7.4.1 Aluminium, silicon, vanadium, nickel, iron, sodium, calcium, zinc and phosphorus. Prepare a 250 mg/l working solutions by diluting 25 ml of the 1 000 mg/l standard solution (4.11.1, 4.11.2, 4.11.3, 4.11.4, 4.11.5, 4.11.6, 4.11.7, 4.12.8, 4.12.9) to 100 ml with water. To each of thirty-six clean 100 ml volumetric flasks (5.9), add 0,4 g flux (4.8) and 50 ml of the tartaric acid/hydrochloric acid solution (4.9.1). To fourflasks add 2 ml, 4 ml, 10 ml and 20 ml of the 250 mg/l aluminium working solution and dilute to 100 ml with water. To another four flasks add 2 ml, 4 ml, 10 ml and 20 ml of the 250 mg/l silicon working solution and dilute to 100 ml with water. Continue to make similar solutions for each of the elements.
NOTE 10 - The calibration solutions contain 5 mg/l, 10 mg/l, 25 mg/l and 50 mg/l of aluminium, silicon, vanadium, nickel, iron, sodium, calcium, zinc and phosphorus each element respectively.
7.4.2 Storage. Transfer all standards immediately after preparation to 100 ml plastic bottles (5.15).
NOTE 12 - When aluminium, silicon, vanadium, nickel, iron, sodium, calcium, zinc and phosphorus are being determined together, it is possible to combine the 5 mg/l to 50 mg/l calibration solution, providing that there are no compatibility problems caused by the reagents used in the preparation of standard solutions given in 4.11.1 to 4.11.9, or contaminant levels of the other elements in the range that are likely to cause unacceptable blank levels of the respective elements being determined.
7.5 Setting up the spectrometer
7.5.1 General.Consultandfollowthe manufacturer's instructions for the operation of the inductively coupled plasma emission spectrometer (5.2).
NOTE 13 - Design differences between spectrometers, ICP excitation sources and different analytical wavelengths for individual spectrometers make it impracticable to specify the required manipulations in detail.
7.5.2 Peristaltic pump. If using a peristaltic pump, inspect the pump tubing and replace it, if necessary, before starting each day. Verify the solution uptake rate and adjust it to the desired rate.
7.5.3 ICP excitation source. Ignite the ICP excitation source at least 30 min before performing an analysis. During this warm-up period, nebulize water through the plasma torch.
NOTE 14 - Some manufacturers may recommend even longer warm-up periods.
7.5.4 Wavelength profiling. Perform any wavelength profiling that may be called for in the normal operation of the instrument.
7.5.5 Operation parameters. Assign the appropriate operating parameters to the instrument task file so that it is possible to determine the desired elements. Include the following parameters: element, wavelength, background correction points (optional), inter-element correction factors (optional), integration time and three consecutive repeat integrations.
7.5.6 Calibration curve. Prepare a five-point calibration curve using the blank and working standards at the beginning of analysis of each batch of samples.
When carrying out this test method for the first time, check the linearity of the spectrometer using the blank and working standards. If the response is linear over the analytic range, a two-point (blank plus high standard) or, if preferred, a three-point (blank plus intermediate plus high) calibration is sufficient. If linearity is not achieved, use multiple standards for the calibration curve.
7.5.7 Analysis of the test solution. Analyze the test portions in the same manner as the calibration standards (i.e. same integration time, background correction points, plasma conditions, etc.). Rinse the plasma torch between samples by nebulizing water for 10 s.
If it is found that a test solution gives an element content above that of the highest calibration solution, dilute it in the same matrix, i.e. blank solution (7.3), to bring it within the range of the calibration solutions.
Analyze one of the calibration standards after every fifth test portion. If any element content is outside 5 % of the nominal value, make any adjustments to the instrument that are necessary and repeat the calibration.
8 Calculation
Calculate E, the content of the element of interest as Al, Si, V, Ni, Fe, Na, Ca, Zn or P respectively in milligrams per kilogram from the following equation:
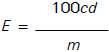
where
c is the element content of interest in mg/l as read from the calibration graph or direct readout;
d is the dilution factor, calculated from the volumes and aliquots taken to produce the test solution;
m is the mass of the test portion, in grams.