

5. General Quality Requirements for the Laboratory
5.1 Establishment and maintenance of a quality system shall include stated objectives in the following areas: a laboratory's adherence to test method requirements, calibration and maintenance practices, and its quality control program. Laboratory quality objectives should encompass the laboratory's continuous improvement goals as well as meeting customer requirements.
5.2 Management shall appoint a representative to implement and maintain the quality system in the laboratory.
5.3 Laboratory management shall review the adequacy of the quality system and the activities of the laboratory for consistency with the stated quality objectives at least annually.
5.4 The quality management system shall have documented processes for:
5.4.1 Sample management (see Section 6),
5.4.2 Data and record management (see Section 7),
5.4.3 Control and implementation of test methods (see Section 8),
5.4.4 Equipment calibration and maintenance (see Section 9),
5.4.5 Quality control (see Section 10),
5.4.6 Audits and proficiency testing (see Section 11),
5.4.7 Corrective and preventive action (see Section 13),
5.4.8 Ensuring that procured services and materials meet the contracted requirements, and
5.4.9 Ensuring that personnel are adequately trained to obtain quality results (see Section 15).
6. Sample Management
6.1 The elements of sample management shall include at a minimum:
6.1.1 Procedures for unique identification of samples submitted to the laboratory.
6.1.2 Criteria for sample acceptance.
6.1.3 Procedures for sample handling.
6.1.4 Procedures for sample storage and retention. Items to consider when creating these procedures include:
6.1.4.1 Applicable government - local, state, or national - regulatory requirements for sample retention period, shelf life, and time-dependent tests that set product stability limits,
6.1.4.2 Type of sample containers required to preserve the sample,
6.1.4.3 Control of access to the retained samples to protect their validity and preserve their original integrity,
6.1.4.4 Storage conditions,
6.1.4.5 Required safety precautions, and
6.1.4.6 Customer requirements.
6.1.5 Procedures for sample disposal in accordance with applicable government regulatory requirements.
NOTE 2 - This may be handled through a separate chemical hygiene or waste disposal plan.
7. Data and Record Management
7.1 Reports of Analysis:
7.1.1 The work carried out by a laboratory shall be covered by a certificate or report that accurately and unambiguously presents the test results and all other relevant information.
NOTE 3 - This report may be an entry in a Laboratory Information Management System (LIMS) or equivalent system.
7.1.2 The following items are suggested for inclusion in laboratory reports:
7.1.2.1 Name and address of the testing laboratory,
7.1.2.2 Unique identification of the report (such as serial number) on each page of the report including version identification if the report has been updated,
NOTE 4 - Occasionally, a report may be updated and a version identification will enable one version of the report to be distinguished from another. This is necessary to determine which report version was the original and which is the most current. Simple conventions such as last updated date are useful means of version identification.
7.1.2.3 Name and address of the customer,
7.1.2.4 Order number,
7.1.2.5 Description and identification of the test sample including comments on the sample condition particularly if it is likely to have an adverse effect on the sample integrity,
7.1.2.6 Date of receipt of the test sample and date(s) of performance of test, as appropriate,
7.1.2.7 Identification of the test specification, method, and procedure,
7.1.2.8 Description of the sampling procedure, where relevant,
7.1.2.9 Any deviations, additions to or exclusions from the specified test requirements, and any other information relevant to a specific test,
7.1.2.10 Disclosure of any nonstandard test method or procedure utilized,
7.1.2.11 Measurements, examinations, and derived results including units of measurement, supported by tables, graphs, sketches, and photographs as appropriate, and any failures identified,
7.1.2.12 Minimum-maximum product specifications, if applicable,
7.1.2.13 Astatement of the measurement uncertainty (where relevant or required by the customer),
7.1.2.14 Any other information which might be required by the customer,
7.1.2.15 A signature and job title of person(s) accepting technical responsibility for the test report and the date of issue, and
7.1.2.16 A statement on the laboratory policy regarding the reproduction of test reports.
7.1.3 Items actually included in laboratory reports should be specified by laboratory management or agreements with customers, or both.
7.1.4 Procedures for corrections or additions to a test report after issue shall be established.
7.2 Reporting and Rounding the Data:
7.2.1 The reporting requirements specified in the test method or procedure shall be used (unless specifically required otherwise by the customer or applicable regulations).
7.2.2 If rounding is performed, the rounding protocol of Practice E29 should be used unless otherwise specified in the method, procedure, or governing specification.
7.3 Records of Calibration and Maintenance:
7.3.1 Procedures shall be established for the management of instrument calibration records. Such records usually indicate the instrument calibrated, method or procedure used for calibration, the dates of last and next calibrations, the person performing the calibration, the values obtained during calibration, permissible tolerances, and the nature and traceability (if applicable) of the calibration standards (that is, certified values). Records may be electronic.
7.3.2 Procedures shall be established for the management of instrument maintenance records. Such records usually indicate the instrument maintained, description of the maintenance performed, the dates of last and next maintenance, and the person performing the maintenance. Records may be electronic.
NOTE 5 - For instruments that require calibration, calibration and maintenance records may be combined.
7.4 Quality Control (QC) Testing Records:
7.4.1 The laboratory shall have documented procedures for creating and maintaining records for analysis of QC samples. It is recommended that such records include the sample name and source, the test(s) for which it is to be used, the assigned values and their uncertainty where applicable, and values obtained upon analysis. Additionally, it is recommended that the receipt date or date put into active QC use in the laboratory be documented, along with the expiration date (if applicable).
7.4.2 Procedures for retaining completed control charts should be established. It is recommended that these records include the date the control charts were changed and the reason for the change.
7.5 Record Retention:
7.5.1 The record system should suit the laboratory's particular circumstances and comply with any existing regulations and customer requirements.
7.5.2 All data shall be maintained according to laboratory, company, customer, or regulatory agency requirements, or a combination thereof.
7.5.3 Procedures for retaining records, including electronic, of all original observations, calculations and derived data, calibration records, and final test reports for an appropriate period shall be established. The records for each test should contain sufficient information to permit satisfactory replication of the test and recalculation of the results.
7.5.4 The records shall be held in a safe and secure storage. A system shall exist that allows locating the required documents in a reasonable period of time.
8. Test Methods
8.1 The laboratory shall have documented test methods and procedures for performing the required tests.
8.2 The test methods that are stated in the product specifications or agreed upon with customers shall be used for sample analysis.
8.3 These test methods shall be maintained up-to-date and be readily available to the laboratory staff.
8.4 The laboratory shall have procedures for the approval, documentation, and reporting of deviations from the test method requirements or the use of alternative methods.
9. Equipment Calibration and Maintenance
9.1 Procedures shall be established to ensure that measuring and testing equipment is calibrated, maintained properly, and is in statistical control. Items to consider when creating these procedures include:
9.1.1 Records of calibration and maintenance (see 7.3),
9.1.2 Calibration and maintenance schedule,
NOTE 6 - The calibration frequency may vary with the instrument type and its frequency of use, some needing calibration before each set of analyses, others requiring calibration at less frequent periods, or triggered by a QC chart out-of-statistical-control situation.
9.1.3 Traceability to national or international standards,
NOTE 7 - Where the concept of traceability to national or international standards of measurement is not applicable, the testing laboratory shall provide satisfactory evidence of test result accuracy (for example, by participation in a program of interlaboratory comparisons).
9.1.4 Requirements of the test method or procedure,
9.1.5 Customer requirements, and
9.1.6 Corrective actions (see Section 13).
9.2 The performance of apparatus and equipment used in the laboratory but not calibrated in that laboratory (that is, pre-calibrated, vendor supplied) should be verified by using a documented, technically valid procedure at periodic intervals.
9.3 Calibration standards shall be appropriate for the method and characterized with the accuracy demanded by the analysis to be performed. Quantitative calibration standards should be prepared from constituents of known purity. Use the primary calibration standards or CRMs specified or allowed in the test method.
9.3.1 Where appropriate, values for reference materials should be produced following the certification protocol used by NIST or other standards issuing bodies, and, should be traceable to national or international standard reference materials, if required or appropriate.
9.3.2 The materials analyzed in proficiency testing programs meeting the requirements of Practice D6300 or ISO 4259 may be used as reference materials, provided no obvious bias or unusual frequency distribution of results are observed. The consensus value is most likely the value closest to the true value of this material; however, the uncertainty attached to this mean value will be dependent on the precision and the total number of the participating laboratories.
9.4 The laboratory shall establish procedures for the storage of reference materials in a manner to ensure their safety, integrity, and protection from contamination (see 6.1.4).
9.5 Records of instrument calibration shall be maintained (see Section 7).
9.6 If an instrument is found to be out of calibration, and the situation cannot be immediately addressed, then the instrument shall be taken out of operation and tagged as such until the situation is corrected (see Section 13).
10. Quality Control
10.1 Quality Control Practices:
10.1.1 Use appropriate quality control charts or other quality control practices (for example, like those described in Practice D6299) for each test method performed by the laboratory unless specifically excluded. Document cases where quality control practices are not employed and include the rationale.
10.1.2 This practice advocates the regular testing of quality control samples with timely interpretation of test results. This practice also advocates using appropriate control charting techniques to ascertain the in-statistical-control status of test methods in terms of precision, bias (if a standard is being used), and method stability over time. For details concerning QC sample requirements and control charting techniques, refer to Practice D6299. The generally accepted practices are outlined in 10.1.3 through 10.4.4.
10.1.3 Test QC samples on a regular schedule. Principal factors to be considered for determining the frequency of testing include: (1) frequency of use of the analytical measurement system, (2) criticality of the parameter being measured and business economics, ( 3) established system stability and precision performance based on historical data, (4) regulatory requirements, (5) contractual provisions, and (6) test method requirements.
10.1.3.1 If site precision for a specific test has not been established as defined by Practice D6299, then the recommended frequency for analysis of QC samples is one QC out of every ten samples analyzed. Alternatively, one QC sample is analyzed each day that samples are analyzed, whichever is more frequent.
10.1.3.2 Once the site precision has been established as defined by Practice D6299, and to ensure similar quality of data is achieved with the documented method, the minimal QC frequency may be adjusted based on the Test Performance Index (TPI) and the Precision Ratio (PR).
10.1.3.3 Table 1 provides recommended minimal QC frequencies as a function of PR and TPI. For those tests, which are performed infrequently, for example less than 25 samples are analyzed monthly, it is recommended that at least one QC sample be analyzed each time samples are analyzed.
10.1.3.4 In many situations, the minimal QC frequency as recommended by Table 1 may not be sufficient to ensure adequate statistical quality control, considering, for example, the significance of use of the results. Hence, it is recommended that the flowchart in Fig. 1 be followed to determine ifa higher QC frequency should be used.
10.1.3.5 The TPI should be recalculated and reviewed at least annually. Adjustments to QC frequency should be made based on the recalculated TPI by following sections 10.1.3.1 and 10.1.3.2.
10.1.4 QC testing frequency, QC samples, and their test values shall be recorded.
10.1.5 All persons who routinely operate the system shall participate in generating QC test data. QC samples should be treated as regular samples.
NOTE 8 - Avoid special treatment of QC samples designed to "get a better result". Special treatment seriously undermines the integrity of precision and bias estimates.
10.1.6 The laboratory may establish random or blind testing, or both, of QC or other known materials.
10.2 Quality Control Sample and Test Data Evaluation:
10.2.1 QC samples should be stable and homogeneous materials having physical or chemical properties, or both, representative of the actual samples being analyzed by the test method. This material shall be well-characterized for the analyses of interest, available in sufficient quantities, have concentration values that are within the calibration range of the test method, and reflect the most common values tested by the laboratory. For QC testing that is strictly for monitoring the test method stability and precision, the QC sample expected value is the control chart centerline, established using data obtained under site precision conditions. For regular QC testing that is intended to assess test method bias, RMs, or CRMs with independently assigned ARVs should be used. The results should be assessed in accordance with Practice D6299 requirements for check standard testing. For infrequent QC testing for bias assessment, refer to Practice D6617.
NOTE 9 - It is not advisable to use the same sample for both a calibrant and a QC sample. It is not advisable to use the same chemical lot number for both a calibrant and a QC sample.
10.2.2 If the QC material is observed to be degrading or changing in physical or chemical characteristics, this shall be immediately investigated and, if necessary, a replacement QC material shall be prepared for use.
NOTE 10 - In a customer-supplier quality dispute, it may be beneficial to provide the customer with the laboratory's test results on QC material to demonstrate testing proficiency. Practice D3244 may be useful.
10.3 Quality Control Charts:
10.3.1 QC sample test data should be promptly plotted on a control chart and evaluated to determine if the results obtained are within the method specifications and laboratory-established control limits. The charts used should be appropriate for the testing conditions and statistical objectives. Corrective action should be taken and documented for any analyses that are out-of-control (see Section 13).
NOTE 11 - Charts such as individual, moving average and moving range, exponentially weighted moving average, or cumulative summation charts may be used as appropriate. Refer to Practice D6299 for guidance on plotting these charts.
10.3.1.1 The charts should indicate the test method, date when the QC analyses were performed, and who performed them. Test samples should not be analyzed or results for samples should not be reported until the corresponding QC data are assessed and the testing process is verified to be in statistical control. (See 10.1.)
10.3.2 Adequate training should be given to the analysts to enable them to generate and interpret the charts.
10.3.3 It is suggested that the charts be displayed prominently near the analysis workstation, so that all can view and, if necessary, help in improving the analyses.
10.3.4 Supervisory and technical personnel should periodically review the QC charts.
10.3.5 The laboratory should establish written procedures outlining the appropriate interpretation of QC charts and responses to out-of-statistical-control situations observed.
10.3.5.1 When an out-of-statistical-control situation has been identified, remedial action should be taken before analyzing further samples. In all such cases, run the QC sample and ensure that a satisfactory result can be obtained before analyzing unknown samples.
NOTE 12 - A generic checklist for investigating the root cause of unsatisfactory analytical performance is given in Appendix X1.
10.3.6 Out-of-control situations may be detected by one or more analyses. In these cases, it may be necessary to retest samples analyzed during the period between the last in-control QC data point and the QC data point that triggered the out-of-statistical-control notice (or event) using retained samples and equipment known to be in control. If the new analysis shows a difference that is statistically different from the original results, and the difference exceeds the established site precision of that test, the laboratory should decide on what further actions are necessary (see Section 13).
10.4 Revision of Control Charts - QC chart revision is covered in detail in Practice D6299. Control charts shall be revised only when the existing limits are no longer appropriate. As a guideline, revisions may be needed when:
10.4.1 Additional information becomes available,
10.4.2 The process has improved,
10.4.3 A new QC material is initiated and the mean value is different than the previous QC material, or
10.4.4 There are major changes to the test procedure.
11. Audits and Proficiency Testing
11.1 Audits:
11.1.1 A laboratory shall have a system to periodically review its own practices to confirm continued conformance to the laboratory's documented quality system. Even if the laboratory is subjected to a formal external audit (for example, as a requirement of ANSI/ISO/ASQ Q9000), it is important to have internal audits since the internal reviewers may be more familiar with their laboratory's requirements than the external auditors.
11.1.2 Audits of test methods should be conducted to confirm adherence to the documented test methods. The performance of the entire test should be observed and checked against the official specified test method. An annual audit of test methods is recommended.
NOTE 13 - These audits may be part of the quality system audits or may be separate.
11.1.3 Audit results shall be promptly documented. The team shall report the audit results to management having the authority and responsibility to take corrective action and to its management.
11.1.4 The findings and recommendations of these internal audits shall be reviewed by the laboratory management and acted upon to correct the deficiencies or nonconformances.
11.1.5 The effectiveness of any corrective actions taken in response to an audit shall be verified. The follow-up results shall be documented as required by the quality system procedures or laboratory policy, or both.
11.2 Proficiency Testing:
11.2.1 Regular participation in interlaboratory proficiency testing programs, where appropriate samples are tested by multiple test facilities using a specified test protocol, shall be integrated into the laboratory's quality control program. Proficiency test programs should be used as appropriate by the laboratory to demonstrate testing proficiency relative to other industry laboratories.
NOTE 14 - Document the rationale for not participating in a proficiency test program.
11.2.2 The laboratory shall establish criteria for guiding their participation in interlaboratory testing programs. Such criteria may include factors such as the frequency of use of the target test method, the critical nature of how the customer uses the data, and regulatory considerations. Participation in proficiency test programs can provide a cost-effective alternative to regular CRM testing.
11.2.3 Participants may plot their deviations from the consensus values established by the proficiency test program averages on a control chart to ascertain if their measurement processes are non-biased. The precision of these exchange performance data can also be assessed against precision established by in-house QC sample testing for consistency (see Practice D6299 for details).
11.2.4 Additional guidance related to the analysis and interpretation of proficiency test program results is explained in Guide D7372.
11.2.5 Participation in proficiency testing shall not be considered as a substitute for in-house quality control, as described in 10.1, and vice versa.
12. Test Method Precision Performance Assessment
12.1 The test performance index (TPI) can be used to compare the precision of the laboratory measurements with the published reproducibility of a standard test method. The term TPI is defined as:
test performance index = test method reproducibility/site precision
NOTE 15 - The ASTM International Committee D02 sponsored Interlaboratory Crosscheck Program employs a test performance index based on the ratio of the published ASTM reproducibility to the Robust Reproducibility calculated from the program data. This index is termed the TPI (Industry) to distinguish from the definition in 12.1.
12.2 A precision ratio (PR) is determined for a given published test method so that the appropriate action criteria may be applied for a laboratory's TPI. The PR for a published test method estimates the influence that non-site specific variations has on the published precision. The PR can be calculated by dividing the test method's Reproducibility by the repeatability as shown in Eq 2.
Precision Ratio, PR5 = Test Method reproducibility (R)/Test Method repeatability (r)
where the ratio of R/r is calculated to the nearest integer (that is, 1, 2, 3, 4, …).
12.2.1 A test method with PR greater than or equal to 4, for the purpose of this practice, is deemed to exhibit a significant difference between repeatability and reproducibility. For further explanation on why the greater than or equal to 4 criterion was chosen, please see Appendix X3.
12.3 A laboratory's TPI may be a function of the sample type being analyzed and variations associated with that laboratory. As general guidelines Table 2 may be used once the TPI of that laboratory and the PR of the published standard test method has been calculated. Similar information to that provided in Table 2 is provided in 12.3.1 through 12.3.2.3.
12.3.1 For a published standard test method with a PR less than 4 the following TPI criteria should be applied.
12.3.1.1 A TPI greater than 1.2 indicates that the performance is probably satisfactory relative to ASTM published precision.
12.3.1.2 A TPI greater than or equal to 0.8 and less than or equal to 1.2 indicated performance may be marginal and the laboratory should consider method review for improvement.
12.3.1.3 A TPI less than 0.8 suggests that the method as practiced at this site is not consistent with the ASTM published precision. Either laboratory method performance improvement is required, or ASTM published precision does not reflect achievable precision. Existing interlaboratory exchange performance (if available) should be reviewed to determine if the latter is plausible.
12.3.2 For a published standard test method with a PR greater than or equal to 4 the following TPI criteria should be applied.
12.3.2.1 A TPI greater than 2.4 indicates that the performance is probably satisfactory relative to ASTM published precision.
12.3.2.2 A TPI greater than or equal to 1.6 and less than or equal to 2.4 indicated performance may be marginal and the laboratory should consider method review for improvement.
12.3.2.3 A TPI less than 1.6 suggests that the method as practiced at this site is not consistent with the ASTM published precision. Either laboratory method performance improvement is required, or ASTM published precision does not reflect precision achievable. Existing interlaboratory exchange performance (if available) should be reviewed to determine if the latter is plausible.
12.3.3 A laboratory may choose to set other benchmarks for TPI, keeping in mind that site precision of an adequately performing laboratory cannot, in the long run, exceed the practically achievable reproducibility of the method when PR is less than 4 or approaches repeatability when PR is much greater than 4.
NOTE 16 - Experience has shown, for some methods, published reproducibility is not in good agreement with the precision achieved by participants in well-managed crosscheck programs. Users should consider this fact when evaluating laboratory performance using TPI.
12.4 A laboratory should review their precision obtained for multiple analyses on the same sample. The site precision of the QC samples can be compared with the reproducibility or repeatability given in the standard test methods to indicate how well a laboratory is performing against the industry standards.
12.5 A laboratory precision significantly worse than the published test method reproducibility may indicate poor performance. An investigation should be launched to determine the root cause for this performance so that corrective action can be undertaken if necessary. Such a periodic review is a key feature of a laboratory's continuous improvement program.
13. Corrective and Preventive Action
13.1 The need for corrective and preventive action may be indicated by one or more of the following unacceptable situations:
13.1.1 Equipment out of calibration,
13.1.2 QC or check sample result out of control,
13.1.3 Test method performance by the laboratory does not meet performance criteria (for example, precision, bias, and the like) documented in the test method,
13.1.4 Product, material, or process out of specification data,
13.1.5 Outlier or unacceptable trend in an interlaboratory cross-check program,
13.1.6 Nonconformance identified in an external or internal audit,
13.1.7 Nonconformance identified during review of laboratory data or records,
13.1.8 Customer complaint.
13.2 When any of these situations occur, the root cause should be investigated and identified. Procedures for investigating root cause should be established. Items to consider when creating these procedures include:
13.2.1 Determining when the test of equipment was last known to be in control,
13.2.2 Identifying results that may have been adversely affected,
13.2.3 How to handle affected results already reported to a customer,
13.2.4 What to do if the root cause cannot be determined, and
13.2.5 What to do if it is determined that the original data is correct.
13.2.6 It is possible that the analytical results are correct, even if they don't meet specifications. Procedures should consider this possibility. See Appendix X1 for a checklist for investigating the root cause of unsatisfactory analytical performance.
13.3 Procedures should also be established for the identification and implementation of appropriate corrective and preventive action so that the situation does not reoccur. This may involve:
13.3.1 Training or retraining personnel,
13.3.2 Reviewing customer specifications,
13.3.3 Reviewing test methods and procedures,
13.3.4 Establishing new or revised procedures,
13.3.5 Instrument maintenance and repair,
13.3.6 Re-preparation of reagents and standards,
13.3.7 Recalibration of equipment,
13.3.8 Re-analysis of samples, and
13.3.9 Additional QC sample analysis.
13.3.10 The situation, root cause, and corrective/preventive action taken should be documented promptly. A corrective and preventive action report is a suitable format for documentation.
13.3.11 The report should be reviewed and approved by management and then verified for effectiveness of corrective/preventive action.
13.4 Quality control charts (see 10.3) are a method of preventive action and should be evaluated on a regular basis to prevent, when possible, out-of-statistical-control situations.
14. Customer Complaints
14.1 A procedure shall exist to follow-up on customer complaints or non-conformances brought to the laboratory's attention by a client. The result of such investigation should be communicated to the customer as soon as practical.
15. Training
15.1 Laboratory management shall ensure that all staff performing testing or interpreting data, or both, are appropriately trained.
15.2 Laboratory training should cover at a minimum the following areas: safety, test methods, and company policies and procedures. Training is specifically required as specified in: 5.4.9, 10.3.2, 13.3, and X1.1.12.
15.3 Records of training should be maintained.
16. Relationship with Other Quality Standards
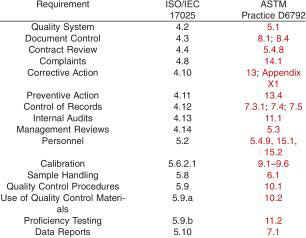
16.1 Some laboratories in the petrochemicals testing area have been registered to ISO/IEC 17025. There are a number of similarities between the ISO standard and this practice in key areas of managing laboratory quality. For example:
16.2 Measurement Uncertainty - For test methods under the jurisdiction of Committee D02, measurement uncertainty as required in ISO/IEC 17025, as practiced by a laboratory, can be estimated by multiplying 2x the site precision standard deviation as defined in Practice D6299.
NOTE 17 - The complexity and empirical nature of the majority of D02 methods preclude the application of rigorous measurement uncertainty algorithms. In many cases, interactions between the test method variables and the measurand cannot be reasonably estimated due to the covariance of the variables that affect the measurand. The site precision approach estimates the combined effects of these variables on the total uncertainty for the measurand.
NOTE 18 - The methodology of using site precision established using QC materials and control charts to estimate measurement uncertainty assumes that the laboratory is unbiased. This assumption should be validated periodically using check standards. See Practice D6617 or Practice D6299 for further guidance.
17. Keywords
17.1 audit; calibration; control charts; proficiency testing; quality assurance; quality control; test performance index